De-risking the Transition from Vial to Drug-Device Combination Product
The combination product market has seen a significant amount of growth,i driven primarily by the rise in chronic disease indications, the demands for self-administered therapies and technological advancements. The use of combination products simplifies the process of drug administration for the patient and/or the caregiver and, in some cases, allows the patient to receive treatment at home rather than having to travel to a medical facility. However, there are many challenges that need to be navigated as one considers the transition from a vial system to a needle-based combination product.
The US FDA defines a combination product as comprising two or more constituents and classifies them into four types:
- Drug + Device
- Biologic + Device
- Drug + Biologic
- Drug + Biologic + Device
Each constituent retains its regulatory status. For example, a combination product comprised of a drug or biologic and a device must meet the regulatory requirements of both the drug or biologic and the device, with the filing center being based on the primary mode of actionii of the combination product, either CDERiii/CBERiv or CDRH.v
In the EU, a combination productvi can also be a medicinal substance with an integral or non-integral , (co-packaged or referenced) medical device and the regulatory filing must include both the medicinal substance filing and the device filing, as discussed below.
This blog primarily addresses combination products classified as Single Entity or Co-Packaged where the applicant either 1) has a drug or biologic packaged in a vial and seeks to expand to a combination product or 2) has a drug or biologic that they are considering whether to package in a vial or in a combination product. In these cases, the primary mode of action is the drug or biologic and the filing goes through CDER or CBER, respectively. For the EU, these would be either a medicinal substance with an integral medical device (Single-Entity analog) and filed through the European Medicines Agency (EMA) with either a Notified Body opinion or a CE Mark for the device or alternatively two separate filings, in the case of non-integral products, one to the EMA for the medicinal constituent and a second to a Notified Body for the device constituent to receive a CE mark.
Combination Product Development Challenges
An important part of combination product development is compiling the regulatory submission. Without approval from regulatory bodies, products do not make it into regulated markets. The regulatory landscape is ever-changing within the combination product development space and regulators now have a lot of direct combination product subject matter expertise. As a result, there is an expectation from regulators that pharma companies that file a combination product application have a full understanding of the drug/biologic constituent as well as an equally full understanding of the device constituent, which is not their forte.
The second challenge is that formulations and therapies are increasingly complex and drug stability can be highly dependent on its interaction – or hopefully lack of interaction - with the primary container and with its flow path. As such, the compatibility and performance of the related system critical components used with the drug/biologic constituent must be understood. Unlike vial systems, combination products have moving parts and often require lubrication or other modifications that can affect drug stability.
The third challenge is the need to deliver a great patient experience. The growing preference for self- or home-administration means that more medications are integrated with devices. However, devices must be easy to use and empower patients while addressing costs. This drives demand for an improved patient, or user, experience that may require an advanced drug delivery system.
And finally, with more competition among drugs treating the same disease, as well as the growth of generics and biosimilars, there is a greater need for product differentiation which can be achieved through an improved drug delivery system.
Combination Product Regulatory Requirements
21 CFR part 4viiilists the requirements for a combination product and there are a number of issued US FDA guidance documents and draft guidance documents that elaborate on the possible paths to take for a dossier submission. The preferred approach, when considering converting a vial packaged drug or biologic to a combination product is to follow the cGMPs for the drug (21 CFR parts 210ix and 211x) or the biologic (21 CFR parts 600 through 680xi) and then also add device quality system regulation provisions from 21 CFR part 820. xii This is what the FDA calls a “streamlined approach.“xii These device provisions are also listed in ISO 13485:2016.xiv
The Design Controls section (21 CFR part 820.30xv) has a number of subheadings including Design Input, where the user requirements are specified, Design Verification, where performance testing is executed to show that the Design Outputs match the Design Inputs (or that the product does what it should do) and Design Validation, where Human Factor studies are typically used to show that the device meets defined user needs and intended uses under actual or simulated use conditions.
Meanwhile, in the EU, MDRxvi Article 117 describes approaches for a medicinal substance with an integral medical device, which in the US would constitute single entity combination products. Co-packaged and cross-labelled devices are not mentioned; in those scenarios, the device component must have its own CE mark and the medicinal product must have its own Competent Authority approval, as mentioned above. MDR Article 117 proposes two options for the device constituent of single entity combination products: one can either file for a CE mark for the device-constituent or obtain a Notified Body Opinion on the conformity of the device to MDR requirements.xvii The Notified Body Opinion requires Technical Documentation, as described in Annex II of the MDR, which includes the General Safety and Performance Requirements (GSPR). The components of the combination product (i.e., seals, plungers, barrels, etc.) must be assessed appropriately with supporting data covering aspects around properties, compatibility and contaminants. Having the component-supplier provide this information would be very helpful for the regulatory filing.
A focused approach to move from a vial format to a device driven combination product is recommended with three critical de-risk factors which include: the Patient, the Plan and the Product.
The Patient
A proactive approach to obtaining first pass regulatory approvals must first de-risk the patient, who must always be the primary focus of the combination product. For example, as therapies become more complex and shift towards home administration, pharma companies are under pressure to bring more intricate devices to market that are also easy for patients to use.
However, this is not easy to achieve with constantly changing regulations and expectations that place more responsibility on pharma companies to prove that not only is their drug constituent safe and effective, which is their expertise, but to also show that the device constituent is safe, effective, reliable and with adequately designed-in human factors consideration, which are not facets of typical pharma company expertise.
Such systems for home administration must involve minimal thought for use. If instructions are unclear or confusing, or if the system has not been thoroughly tested for real world use, including potential misuse, this can result in a poor patient experience, patient pain, promote patient adherence challenges, compromise the therapy and impact overall market acceptance; possibly even face recall. Some of these outcomes can be a direct result of poor component selection, as discussed below, and must be thoroughly explored in Design Verification and in the Human Factors studies performed for Design Validation. The patients and their ability to use the device must be considered at the start of any combination product project.
The PlanSome companies first launch the product in the simplest presentation possible and only once a commercial success is seen do they initiate Lifecycle Management to a more advanced presentation. This is a risky approach since often these companies will find out too late that their product will be at a competitive disadvantage as they initiate Lifecycle Management efforts too late. The results are higher overall costs, unfulfilled market potential and (possibly) loss of momentum to being the first on the market. Biosimilar and generic companies might underestimate the relentlessness of the originators to protect their brands and could be surprised to find that by the time they get their approval to launch the biosimilar or generic, the originator already converted the market to an advanced presentation and the biosimilar or generic is left chasing the innovator.
Pharma companies should plan to start stability studies early on, using real-time and accelerated conditions. If drug or biologic stability will be affected by the new presentation, one would want to know sooner than later.
It is important to partner with suppliers tthat have component and system knowledge and a proven track record in successful regulatory submissions to help minimize surprises. The partner should have meaningful platform data, such as component extractables and device performance data packages, that could be leveraged into device development to save time and resources. Furthermore, it is helpful to partner with industry subject matter experts to establish risk assessment linkages from components to the combination product. These linkages make it easier to avoid surprises, shorten timelines, improve operational efficiencies that reduce costs and increase the probability of regulatory success.
All of this means that the combination product can be launched faster and with a competitive advantage, serve diverse market needs and deliver a better patient experience which would lead to improved adherence to the pharmaceutical regimen.
The Product
De-risking the product begins with a thorough understanding of the user requirements to know exactly what the product needs to do. Based on that, the component manufacturers’ in-depth component knowledge regarding the materials’ chemical and physical interactions and the system integrator’s ability to navigate through proper component selection will result in an optimized device design. This can best be achieved by having a robust supply chain with experienced partners.
Testing StrategyThe combination product regulatory filing will require extensive laboratory testing for Design Verification. The testing will be based on risk assessments and consensus standards. If the pharma company chooses to outsource this activity, the contract laboratory should have the experience and knowledge to perform the necessary testing that provides fit for use data for the combination product from compendial single component requirements to Extractables and Leachables, Container-Closure Integrity, Design Verification, Essential Performance Requirements assessments and Particle testing to confirm that the combination product protects the drug product, performs as intended and provides the best patient experience. Those capabilities are necessary to design a testing program and assess the product from development through qualification, stability and release testing.
Consider the device performance risk assessments and Design Verification Testing of a prefilled syringe inside of an auto-injector (Figure 1). There is the syringe sub-system and the auto-injector system. Each has its own unique functions that need to be fully assessed regarding the impact on patient safety and device effectiveness. For the syringe sub-system, there are many component interactions to understand when it comes to assessing suitability. It is very important to note that it is the elastomeric components that tend to have the most interfaces and interactions and the most correlation to patient safety, device effectiveness and device ease-of-use. Syringe barrel-plunger physical compatibility in the auto-injector affect patient therapy characteristics such as activation, dose delivery time and dose accuracy. When determining holistic suitability, it is important to identify critical functions, especially clinically relevant ones (Essential Performance Requirements) impacting risk severity, where system integration aspects must be fully understood and optimized. .
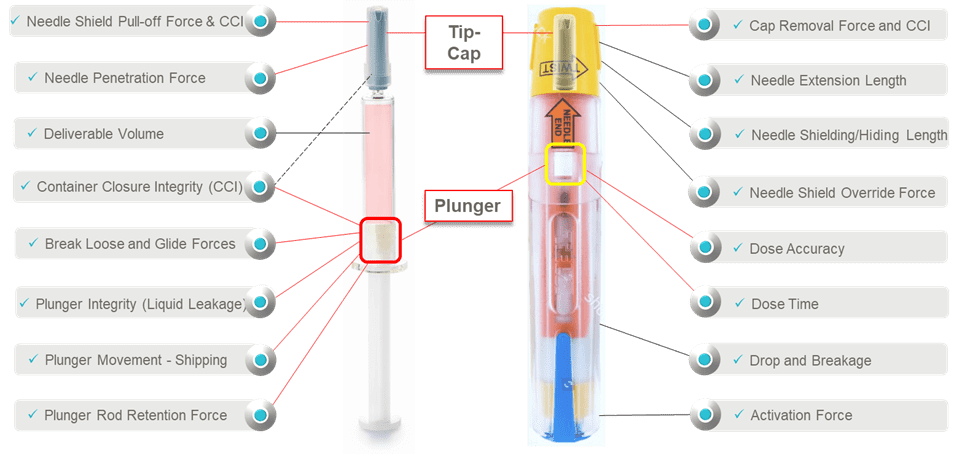
Figure 1. Pre-filled Syringe and Auto-Injector shown along with potential areas for performance testing
Pharmacopeial chapters and consensus standards which cover relevant testing are a good place to start in assessing device performance. These include testing at the component level (i.e., stoppers,xviii, xix seals,xviii and glassxx) and testing at the system level (i.e., container closure integrity,xxi leachablesxxii and overall filled-device performancexix, xxvi, xxvii).
For example, the ISO 11040 series of standards addresses, with mapped-out normative references, all the components of the pre-filled syringe (barrel,xxiii plunger,xxiv cap,xxiii Luer,xxiii nestxxv) as well as the system as a whole,xxvi which is critical to demonstrate that the drug delivery system is fit for purpose. Meanwhile, ISO 11608-5xxvii addresses the needs for evaluating auto-injector performance.
When developing the component, device and combination product risk management strategies, ISO standards are particularly insightful when in risk discovery mode. The normative references therein are helpful in understanding overall device-component and individual component-to-component relationships. It is important to proactively and scientifically discover and address risks early in the development of a component, device, or process to allow oneself the opportunity to make the necessary changes.
A holistic functional suitability and fitness for intended use assessment must include other factors that will have a direct influence on the combination product system. It is important to understand system performance beyond the basics and this should, for example, include aspects related to fill-finish, environmental conditions for use and storage as well as distribution cycles. xxviii
In summary, when transitioning from a vial to a combination product system, the best approach is to have a holistic de-risk strategy with an intentional focus on the Patient, the Plan and the Product. It is vital to select partners with in-depth understanding of the evolving regulatory landscape, that have a systems mindset, that have industry partnerships and that have subject matter expertise in areas required to establish drug-device suitability. With this approach, it is easier to avoid surprises, shorten timelines, improve operational efficiencies that reduce costs, increase the probability of regulatory success, and ultimately reduce risk to the patient. This approach will get the combination product launched and into the market faster while delivering a safe, effective, and positive patient experience.
Acknowledgment
This narrative is based on seminars that Jennifer Riter, Daniel Bantz, Marc Uerdingen and Tomer El-Gad have presented on this topic.
References
i. FY2019 Office of Combination Products Performance Report to Congress as required by the Medical Device User Fee and Modernization Act of 2002. Available at: https://www.fda.gov/media/144887/download. Accessed May 2023.
ii. The primary mode of action is defined as “the single mode of action of a combination product that provides the most important therapeutic action of the combination product,” according to 21 CFR 3.2.
iii. Center for Drug Evaluation and Research at the US FDA. https://www.fda.gov/about-fda/fda-organization/center-drug-evaluation-and-research-cder
iv. Center for Biologics Evaluation and Research at the US FDA. https://www.fda.gov/about-fda/fda-organization/center-biologics-evaluation-and-research-cber
v. Center for Devices and Radiological Health at the US FDA. https://www.fda.gov/about-fda/fda-organization/center-devices-and-radiological-health
vi. Guideline on quality documentation for medicinal products when used with a medical device. EMA/CHMP/QWP/BWP/259165/2019 Accessed July 2023.
vii. 21 CFR 3.2. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-3/subpart-A/section-3.2. Accessed May 2023.
viii. 21 CFR 4. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-4. Accessed May 2023.
ix. 21 CFR 210. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-210. Accessed May 2023.
x. 21 CFR 211. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211. Accessed May 2023.
xi. 21 CFR 600-680. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-F. Accessed May 2023.
xii. 21 CFR 820. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820. Accessed May 2023.
xiii. Current Good Manufacturing Practice Requirements for Combination Products. Guidance for Industry and FDA Staff. Office of Combination Products, Food and Drug Administration. Jan. 2017. Available at: https://www.fda.gov/media/90425/download. Accessed May 2023.
xiv. International Organization for Standardization. (2016) Medical devices — Quality management systems — Requirements for regulatory purposes. (ISO Standard No. 13485:2016). https://www.iso.org/standard/59752.html
xv. 21 CFR 820.30. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820.30. Accessed May 2023.
xvi. Medical Device Regulation, Regulation (EU) 2017/745. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745. Accessed May 2023.
xvii. A Notified Body Opinion is needed regarding the conformity of devices Class II or greater to MDR requirements. Class I devices do not require a Notified Body Opinion letter because they do not require a conformity assessment prior to CE mark.
xviii. USP. Elastomeric Components in Injectable Pharmaceutical Product Packaging/Delivery Systems <381>. In: USP–NF. Rockville, MD: United States Pharmacopeia.
xix. USP. Elastomeric Component Functional Suitability in Parenteral Product Packaging/Delivery Systems <382>. In: USP–NF. Rockville, MD: United States Pharmacopeia. To be official on 01-Dec-2025.
xx. USP. Containers - Glass <660>. In: USP–NF. Rockville, MD: United States Pharmacopeia.
xxi. USP. Package Integrity Evaluation—Sterile Products <1207>. In: USP–NF. Rockville, MD: United States Pharmacopeia.
xxii. USP. Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems <1664>. In: USP–NF. Rockville, MD: United States Pharmacopeia.
xxiii. International Organization for Standardization. (2015). Prefilled syringes – Part 4: Glass Barrels for injectables and sterilized subassembled syringes ready for filling (ISO Standard No. 11040-4:2015). https://www.iso.org/standard/58079.html
and International Organization for Standardization. (2019). Prefilled syringes – Part 6: Plastic Barrels for injectables and sterilized subassembled syringes ready for filling (ISO Standard No. 11040-6:2019). https://www.iso.org/standard/69487.html
xxiv. International Organization for Standardization. (2012). Prefilled syringes – Part 5: Plunger stoppers for injectables. (ISO Standard No. 11040-5:2012). https://www.iso.org/standard/53968.html
xxv. International Organization for Standardization. (2015). Prefilled syringes – Part 7: Packaging systems for sterilized subassembled syringes ready for filling. (ISO Standard No. 11040-7:2015). https://www.iso.org/standard/59937.html
xxvi. International Organization for Standardization. (2016). Prefilled syringes – Part 8: Requirements and test methods for finished prefilled syringes. (ISO Standard No. 11040-8:2016). https://www.iso.org/standard/66036.html
xxvii. International Organization for Standardization. (2022). Needle-based injection systems for medical use — Requirements and test methods — Part 5: Automated functions (ISO Standard No. 11608-5:2022). https://www.iso.org/standard/76627.html
xxviii. ASTM Standard D4169, 2022, " Standard Practice for Performance Testing of Shipping Containers and Systems," ASTM International, West Conshohocken, PA, 2022, https://www.astm.org/d4169-22.html.


