Overview – FDA Draft Guidance - Essential Drug Delivery Outputs for Devices Intended to Deliver Drugs and Biologics
For several years, the Food and Drug Administration (FDA) has required manufacturers of combination products, intended for drug delivery, to identify critical performance requirements that would be evaluated and controlled for the lifecycle of the combination product. Previously, these critical performance requirements were called “Essential Performance Requirements (EPRs),” however, a formal definition was never provided by the FDA. A long-awaited guidance was released by the FDA in June 2024 that establishes and defines new terminology and clarifies expectations from the agency related to establishing, evaluating, and controlling Essential Drug Delivery Output (EDDO) requirements. Notably, the guidance does not address all required information for submission of drug/delivery devices but rather focuses on performance related outputs and adjacent processes.

Essential Drug Delivery Output (EDDO)
EDDOs are defined as design outputs necessary to ensure delivery of the intended drug dose to the intended delivery site. They are a subset of the overall design outputs defined in 21 CFR 820.30(d). Delivery is further clarified to include successful product preparation (as applicable), initiation, progression, completion of the dose and needle safety activation. EDDOs are considered essential for the proper functioning of the device, as a whole, to deliver the drug or biologic and therefore it is important to evaluate the performance and quality of the combination on a system level.
EDDOs could be considered analogous to Critical Quality Attributes (CQA), which are defined in ICH Q8 as physical, chemical, biological, or microbiological properties or characteristics that should be within an appropriate limit, range, or distribution to ensure the desired product quality. Identification of CQAs and EDDOs must be aligned and documented between device and drug manufacturers including specifications, and manufacturing controls.
Identification of EDDOs
Since EDDOs are considered a subset of the overall design outputs for the product, the guidance recommends a filtering approach where all design outputs are listed, then refined into those outputs specific to drug delivery and then further refined as system level outputs that are device, not user, dependent to establish the final determination of EDDOs, as illustrated below:
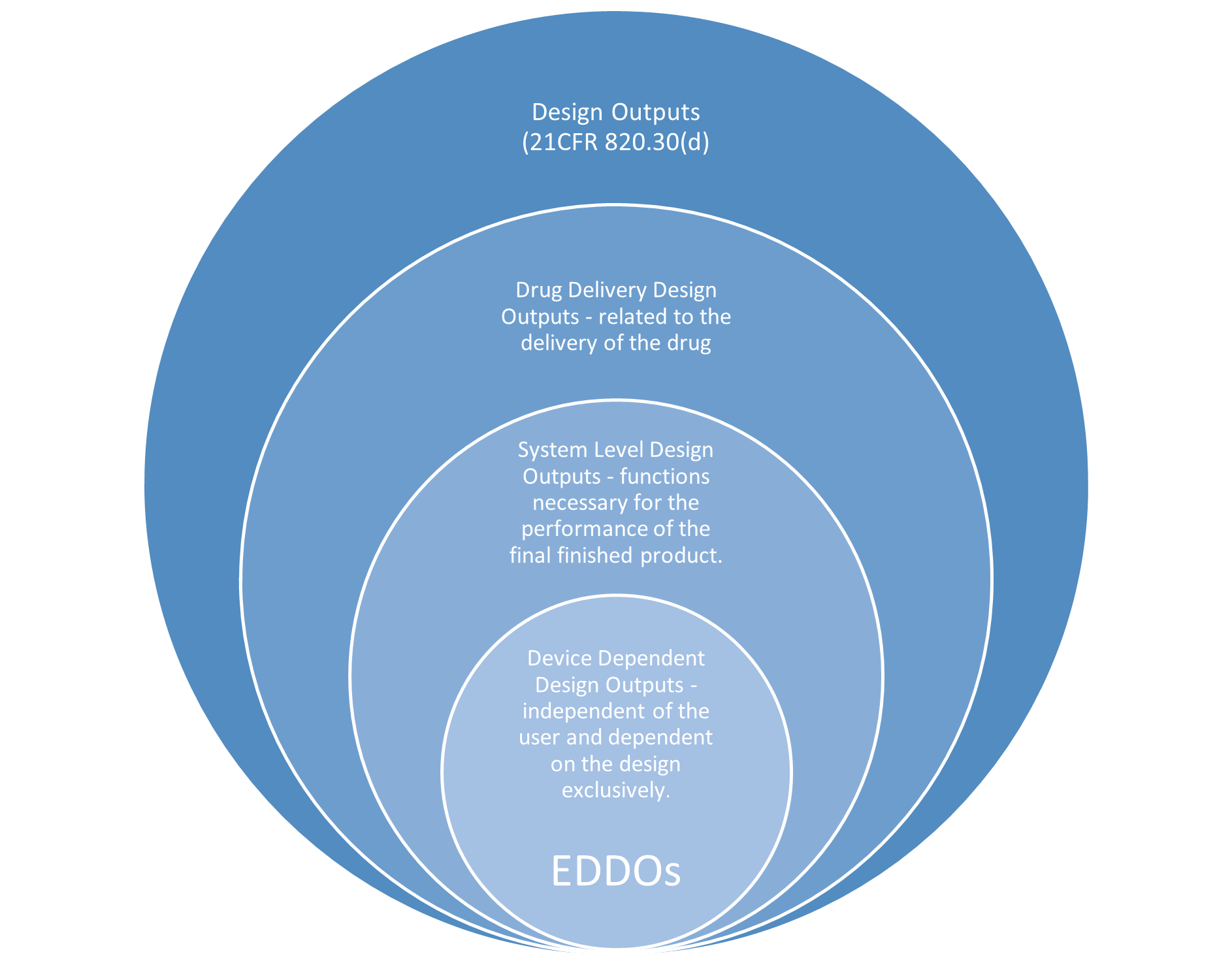
Verification and Validation
Design verification, as described in 21 CFR 820.30(f), is the process used to “confirm that the design output meets the design input requirements,” and is usually based on laboratory testing. Design validation, as described in 21 CFR 820.30(g) is the process used to “ensure that devices conform to defined user needs and intended uses,” and is usually performed through clinical testing. The guidance stresses that all verification and validation activities for EDDOs should be conducted after stress conditioning the product according to those stresses to which the product would be exposed during production, shipping, storage, preparation, and use. Special attention should be paid to all potential environmental conditions including temperature, humidity, pressure, vibration, shock, etc. Risk-based justification for all test conditions (including preconditioning and stress conditions) selected should be included along with the test reports in the submission.Robust risk analysis should also govern sampling plans, taking into account the indication for use, patient population, drug being delivered, context of use and complexity of design and manufacturing. Protocols should include a statistical risk-based sampling plan, where products with a higher risk profile require a larger sample size, and tested product should be representative of the commercial product (manufacturing process and materials).
Shelf-life should be established based on testing conducted using the final finished product, after exposure to any preconditioning deemed applicable, demonstrating that the EDDO is maintained. Accelerated aging may be used to determine shelf life but data should be confirmed by real time aging. Notably, shelf-life evaluation of EDDOs that would not change over time (e.g., physical dimensions such as needle length) would not be required.
Control Strategies
A control strategy is used to ensure that each lot of the final finished product is manufactured to conform to the design outputs. For the purposes of the guidance, the final finished product is considered the product intended for marketing and submitted in the marketing application. Multiple upstream and downstream control processes may require implementation depending on the results of a risk-based assessment for each EDDO. It is important to note how various process steps can influence each other and ultimately the EDDO. Therefore, manufacturers should identify all current controls at all process steps and then evaluate both their effectiveness and their impact to EDDOs. The control strategy should then feed into the risk management processes to determine if sufficient controls are in place or whether additional controls are needed. Information on the overall control strategy, with rationale and risk-based justification, should be contained within the applicant’s submission.
Comparison to other “Performance Requirements”
Although not directly addressed in the draft guidance, additional applicable performance requirements may be needed in the regulatory submission based on the scope of the drug delivery device. These requirements include Primary Functions (as defined in ISO 11608:2022), Essential Performance (as defined in IEC 60601-1) and Established Conditions (as defined in ICH Q12). The applicability of each must be determined by the manufacturer and, unless justified, must be included in the submission in addition to the EDDOs.
| Primary Functions (PFs) | Essential Performance (EPs) | Established Conditions (ECs) |
|---|
| Definitions | Defined in ISO 11608:2022 as thefunction or operation of the needle-based injection system which, if it does not perform tospecifications during use, would directly result in a failure to accurately deliver themedicinalproduct via the correct route and/or directly result in unacceptable harm to the patient | Defined in IEC 60601 as theperformance of a clinical function, other than that related to basic safety, where loss ordegradation beyond the limits specified by the manufacturer results in an unacceptable risk | Defined in ICH Q12 aslegally binding information considered necessary to assure product quality |
| Applicability | PF determination is required for all needle-based injection systems within the stated scopeofISO 11608-1:2022 | EP determination is required for all medical electrical equipment within the stated scope ofIEC60601 | EC determination is required of the application holder for drug substances, products, andDrug-device combinations where the primary mode of action is the drug or biologic. |
| Comparison to EDDOs | PFs are utilized primarily during development to ensure robust verification testing and areincluded as objective evidence in the submission. PFs may be used to generate new data forapplicable post-market changes. | EPs are utilized primarily during development to ensure robust verification testing and areincluded as objective evidence in the submission. EPs may be used to generate new data forapplicable post-market changes. | ECs are utilized primarily in lifecycle management to define the pathway for post-marketchange.ECs are established for both the drug and the device. |
| Regulatory Expectations | Justification for selection, with objective evidence of primary functions is expectedin addition toEDDOs for US regulatory submissions | Justification for selection, with objective evidence of essential performance is expectedin addition toEDDOs for US regulatory submissions | Justification for selection and details of control strategies are required for each EC. ECsarea combination of Critical Quality Attributes (CQAs) (Drug) and EDDOs (Device). |
Impact to CDER and CBER Submissions
Regulatory filings previously submitted for a combination product with information related to Essential Performance Requirements are still valid. However, all documentation referencing old terminology that is not in alignment with the draft guidance should be reviewed to determine the impact and whether updates are needed. Special attention should be made to documentation bridging from clinical to commercial submissions and whether any definitions used previously still reflect alignment with the updated guidance.
Manufacturers should also review the guidance criteria against any current product requirements to ensure alignment. Specifically, for any EDDOs that are not in alignment with the recommendations or examples in the guidance, scientific justification must be provided.
Current Status
The guidance is currently in a draft form and comments are being solicited until September 30th, 2024. Publication of the final guidance has the potential to take up to several years. The current draft guidance represents the agency’s latest thinking and, therefore, the expectation is that all future submissions are in alignment with the current recommendations based on the draft guidance wording.
Important product and safety information and warnings available at: https://www.westpharma.com/products/self-injection-platforms/smartdose/smartdose-3-5


